Step 5: Viewing alignments
At this point, you should have a file called NA12878.bam containing aligned,
duplicate-marked reads. And we'd like to see how they stack up on the genome.
Viewing alignments with tview
The simplest way to do this is using samtools tview. This is a terminal program which shows you a
simple picture of how the read alignments stack up. Let's try this now by running in the terminal:
samtools tview -p chr19:48441038 NA12878.bam
You should see something a bit like this:
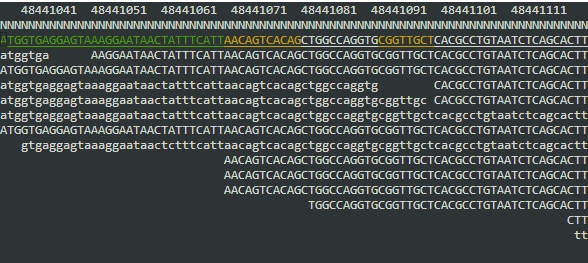
What's shown are the reads as they align to the reference sequence. (Reads that align on the forward strand are in upper case, and reads that aligned to the reverse strand are shown in lower-case.) The numbers along the top are the reference genome position (represented at the leftmost digit; so position 48,441,038 is at the left of the screen).
You can use arrow keys to move around (hold shift to move faster), g to jump to another genome location, or m /
b / n to colour reads by mapping quality, base quality, or nucleotide. (Depending on the reads currently being viewed, it may all stay white....try scrolling around until you see some coloured bases or reads). Press ? for more help. The q key quits the program.
Question. Can you see:
- A sequencing error? (Hint. press
bto colour reads by alignment. The colour scheme can be seen by pressing?) - A SNP or other genetic variant?
- A read with a low-quality alignment? (Hint. press
mto colour reads by mapping quality.)
The answer to the above is 'probably not' at the moment. It becomes much easier when we include the reference
sequence itself. Press q to quit, and now try this command:
samtools tview -p chr19:48441038 NA12878.bam GRCh38_chr19.fa
Now you'll see something a bit more like this:
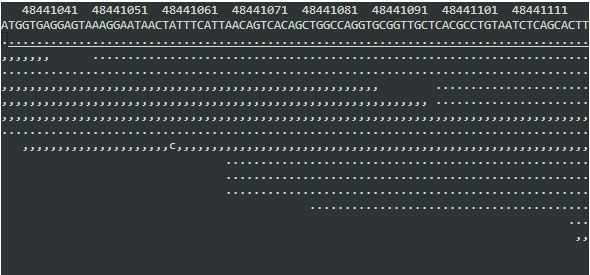
Now dots and commas mean 'reads with the same base as the reference' (dots for forward strand, commas for reverse strand) - anything else means a mismatching base.
Scroll around a bit (remember you can hold <shift> to scroll faster).
Can you find
- a probable sequencing error?
- A SNP or other genetic variant?
- A read with low-quality alignment?
Challenge question
In the command-line tutorial
we looked at the FUT2 gene, which lies on chromosome 19 at 48,695,971-48,705,951.
The SNP rs601338, which is at chr19:48703417 in FUT2, determines
secretor status. Being a non-secretor means that blood group antigens (encoded by ABO and other loci) are not
secreted or expressed into bodily fluids in the normal way. Non-secretors are protected against certain pathogens
including norovirus, but are also more susceptible to others -
so it can be medically important what status you have.
Secretor status is a recessive trait, so the table of genotypes and secretor status at rs601338 is:
| genotype | secretor status |
|---|---|
| GG | secretor |
| GA | secretor |
| AA | non-secretor |
Question What genotype does NA12878 have at rs601338? Are they a secretor or non-secretor?
Next step
Well done! That's the end of the alignment tutorial.
Next, we will go onto looking at some reads in IGV.