Part 2: Genetic association with a case/control trait
Let's fit another logistic regression model to estimate the effect of a genetic variant on an outcome - severe malaria status.
To begin, start an R session and let's load the data:
library(tidyverse)
data = read_tsv( "https://www.chg.ox.ac.uk/bioinformatics/training/msc_gm/2024/data/o_bld_group_data.tsv" )
If the above link doesn't work for any reason, try the this version. (To load this, click 'Raw', copy the URL, and read into R as above.)
What's in the data?
The data contains genotype calls for the genetic variant rs8176719 (and rs8176746), measured in a set of severe malaria cases and population controls from several African countries. These variants lie in the ABO gene and together encode the most common ABO blood group types.
In particular, rs8176719 is the main variant that determines O blood group status. It represents a one base-pair deletion that causes a frameshift and early termination of transcription. If you wish, you can read more about ABO and the O blood group mutation here.
If you want to see what the genetic effect of this variant is, you can look it up in the UCSC genome browser and/or the Ensembl genome browser. For example you should see that rs8176719 is a frameshift variant within ABO gene. (Which exon is it in?). Chromosomes with the deletion allele (like the GRCh38 reference sequence) produce a malformed enzyme that prevents synthesis of A or B antigens by causing a loss of glycosyltransferase activity.
Of course, humans have two copies of chromosome 9, so even if one copy has this mutation, the other one might still express A or B.
The data were collected by seperate case-control studies operating in each country and were genotyped by MalariaGEN. They were described in this paper.
You should look at the data now using the tools you have learnt in R. You should see:
Information on the sample identifier and sex, and ethnicity
The
statuscolumn, which indicates if the sample was collected as a disease case (i.e. came into hospital with severe symptoms of malaria) or a population control.A column with the
rs8176719genotypes (and one with genotypes for another variant,rs8176746.)And some columns marked
PC_1-PC_5.
We'll focus on the status and genotype columns for now.
You should always inspect data closely before starting any analysis.
Do your due diligence on this data frame now. For example
- Which countries are in the data?
- What is the sample size in each country?
- How many samples are severe malaria cases and how many controls?
- What about rs8176719 genotypes?
- Are there any other columns?
- Is there any missing data?
Hint table() or the data verbs can help solve this.
Is O blood group associated with malaria susceptibility?
Let's use logistic regression to test to see if O blood group is associated with malaria status. The basic process is:
- Recode variables, if necessary, to get them in the rifht form
- Use
glm()to fit a logistic regression model - Interpret the parameters
Re-coding variables
An individual has O blood type if they have two copies of the rs8176719 loss-of-function deletion.
So to get started let's create a new variable indicating 'O' blood group status:
# Set to missing first, in case any rs8176719 genotypes are missing
data$o_bld_group = NA
data$o_bld_group[ data$rs8176719 == '-/-' ] = 1
data$o_bld_group[ data$rs8176719 == '-/C' ] = 0
data$o_bld_group[ data$rs8176719 == 'C/C' ] = 0
Or, data verbs style:
data = (
data
%>% mutate(
o_bld_group = c( "C/C" = 0, "-/C" = 0, "-/-" = 1 )[rs8176719]
)
)
Table the genotypes and the new variable, to make sure it's right.
What is the frequency of O blood group in this sample, and in each population?
- Hints and solutions
- Hint
- Table solution
- Data verbs solution
You can get counts of genotypes either using the table() function, or the data verbs.
You then need to use these to compute allele frequencies.
To compute frequencies, you would then need to capture the results to compute frequencies, like this:
counts = table( data$country, data$o_bld_group )
frequencies = counts[,2] / (counts[,1] + counts[,2])
print( frequencies )
(
data
%>% filter( !is.na( o_bld_group ))
%>% group_by( country )
%>% summarise(
total = n(),
o = sum( o_bld_group )
)
%>% mutate( o_frequency = o / total )
)
# A tibble: 5 × 4
country total o o_frequency
<chr> <int> <dbl> <dbl>
1 Cameroon 1182 586 0.496
2 Gambia 5027 2152 0.428
3 Ghana 694 280 0.403
4 Kenya 3054 1546 0.506
5 Tanzania 769 350 0.455
However you do this you should see that blood group O is at about 40-50% frequency in these African populations.
Running the regression
For best results let's also make sure our status variable is coded the right way round, that is, with "CONTROL" as the baseline and "CASE" as the non-baseline outcome:
data$status = factor( data$status, levels = c( "CONTROL", "CASE" ))
Fitting a logistic regression model is now easy:
fit = glm(
status ~ o_bld_group,
data = data,
family = "binomial"
)
This command says:
- fit a logistic regression model...
- modelling the log-odds of severe malaria status in terms of O blood group genotype...
- ...using the given dataframe.
As before, the 'family' argument tells glm that the outcome variable is a binary (0/1) indicator, and that this should be modelled using a binomial distribution and via the log-odds.
Congratulations! You've just fit another logistic regression model.
Interpreting the regression output
The best way to inspect the results of glm() is using the summary() command to get the fitted coefficients:
coefficients = summary(fit)$coefficients
print( coefficients )
You should see something like this:
> print( coefficients )
Estimate Std. Error z value Pr(>|z|)
(Intercept) 0.1489326 0.02630689 5.661354 1.50183e-08
o_bld_group -0.3342393 0.03889801 -8.592709 8.49425e-18
We described how to interpret the parameters output on the warm-up page. Can you use this to answer:
- What do the P-values mean?
Now look at the estimates and try to answer the following questions, first about non-O blood group (baseline) individuals:
- What is the modelled log-odds that a non-O blood group individual is a case?
- What is the modelled probability that a non-O blood group individual is a case in this study?
And the same questions for O blood group individuals:
What is the modelled log-odds that an O blood group individual is a case?
What is the modelled probability that an O blood group individual is a case?
What is the estimated odds ratio?
And the most important question:
- Is O blood group is associated with higher, or lower risk of severe malaria, or neither?
- Hints
- Hint 1
- Hint 2
- Hint 3
The answers on the log-odds scale are provided to you by the fitted regression parameters.
- The baseline log-odds (for non O blood group individuals) is .
- The change in log odds associated with carrying O blood group, or log odds ratio, is .
Add them up to get the log-odds of disease, for an O blood group individual.
To get from log-odds scale to odds scale, transform the estimates using the exponential function.
- The baseline odds of disease (for non-O individuals) is .
- The odds of disease for O individuals is .
Also, the odds ratio is .
If you want to get risk estimates on the probability scale, that's possible too.
However you should remember that in this data, which comes from a case-control study, the risk estimates themselves don't really mean much about the population - they are just about the rates of cases and controls sampled in the study. Nevertheless the (log) odds ratio is still interpretable as an estimate of the difference in risk caused by O blood group. (In fact it's an estimate of relative risk - this is described below for reference, but you can ignore this bit for now.)
Nevertheless, to compute these, you can transform the odds you computed:
Or, you can go all the way from log-odds to probability in one step by using the combined 'logistic' function that we saw earlier:
logistic <- function(x) { exp(x) / (1 + exp(x))}
...which looks like this:
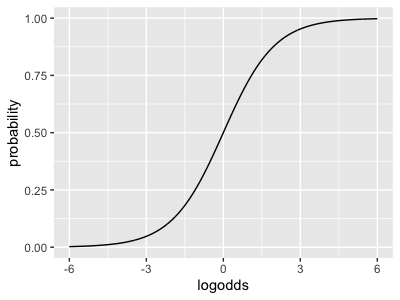
For example, the baseline probability of disease is - and so on.
(This function applies to the log-odds values directly - it doesn't make too much sense to think of the log-odds ratio via this function.)
You should by now have seen that, whichever scale you look on, O blood group is associated with a reduced risk of severe malaria! It has an odds ratio of about , meaning that it's associated with a 28% reduction in risk.
Exploring covariates
Can you re-run the analysis focussing on just one country (say Gambia)? Do you get similar results? What happens to the standard error?
The data has several other variables in it, including information on ethnicity and sex. Can you re-run the analysis including extra variables to see if they affect the results?
Note for variables like ethnicity and the PC_1-PC_5, it makes most sense to do this within one country at a
time. Maybe pick the country with the largest sample size to start with.
Confidence intervals and the forest plot
Let's go back to the overall model fit across all the data:
> print( coefficients )
Estimate Std. Error z value Pr(>|z|)
(Intercept) 0.1489326 0.02630689 5.661354 1.50183e-08
o_bld_group -0.3342393 0.03889801 -8.592709 8.49425e-18
In the warm-up page we described two ways to interpret the standard errors. Let's use the standard errors now to compute confidence/credible intervals.
Computing a confidence/credible internval
The rule to compute a confidence / credible interval is:
Take the regression estimate and go out 1.96 standard errors:
It is often more natural to think on the odds scale. The rule for this is also simple:
- First compute the confidence/credible interval for on the log-odds ratio scale, as above.
- Then use
exp()to convert back to the odds ratio / relative risk scale.
Using the estimated above, compute a 95% confidence interval for the log-odds ratio, and then for the odds ratio now.
- Hints
- Solution
See if you can do this yourself. If you get stuck, see the answer on the tab above.
beta = -0.3342393
se = 0.03889801
lower = beta - 1.96 * se
upper = beta + 1.96 * se
sprintf( " The odds ratio is: %.3f.\n", exp( beta ))
sprintf( "The 95pc interval is: %.3f - %.3f.\n", exp(lower), exp(upper) )
What is your overall interpretation of these regression results?
The forest plot
Ok that's not good enough! Let's make a forest plot of the association evidence across populations. This requires a bit of code which we can implement as follows.
First, rather than typing out our model fit loads of times, let's make a function that fits the regression for us. I'll help myself out by turning the fit parameters into a proper data frame before returning:
fit_regression <- function(
data,
formula = status ~ o_bld_group
) {
library( dplyr )
fit = glm(
formula,
data = data,
family = "binomial"
)
parameters = summary(fit)$coeff # up to here is the same as before
return(
tibble::as_tibble(parameters) # Turn it into a data frame
%>% mutate(
parameter = rownames( parameters ) # Make parameter names into an actual column
)
%>% select( # Rename some columns
parameter,
estimate = Estimate,
se = `Std. Error`,
z = `z value`,
pvalue = `Pr(>|z|)`
)
)
}
If you run this on the original data frame, you should see what it does - gives the same estimates again but with slightly different formatting.
Now let's use some dplyr magic to run seperately in all populations:
estimates = (
data
%>% group_by( country )
%>% reframe(
fit_regression( pick( everything()) )
)
)
You don't have to worry about the details of this code - it is just a way to run our function across each country in the data seperately.
If you want to know how it works, the pick( everything()) bit just says to pass in the whole of the 'current data'
into our function. (Because this is after a group_by(), that means the data for the current group.)
The reframe() function in there is similar to the group by / summarise examples from the data verbs
session, but unlike summarise() allows us to return multiple rows per group. It's handy
for things like this but you don't need to know it for this module.
If you look at the output you should now see two rows per country. One row contains the details for the intercept, and the other the details for the log odds ratio () itself.
Now let's make a forest plot for the effect size parameter. Note the highlighted lines below, which do the actual calculation.
library( ggplot2 )
p = (
ggplot(
data = (
estimates
%>% filter( parameter == 'o_bld_group' )
%>% mutate(
OR = exp( estimate ),
ci_lower_bound = exp( estimate - 1.96 * se ),
ci_upper_bound = exp( estimate + 1.96 * se )
)
)
)
+ geom_segment(
aes(
x = ci_lower_bound,
xend = ci_upper_bound,
y = country,
yend = country
),
colour = 'grey',
linewidth = 1.5 # make it bigger & bolder!
)
+ geom_point(
aes(
x = OR,
y = country,
),
size = 4 # make the points bigger!
)
+ xlim( c( 0.4, 1.2 ))
+ xlab( "Odds ratio and 95% CI" )
+ ylab( "Country" )
# Here's some stuff to make it prettier
+ theme_minimal(16)
+ theme( axis.title.y = element_text( angle = 0, vjust = 0.5 ))
)
print(p)
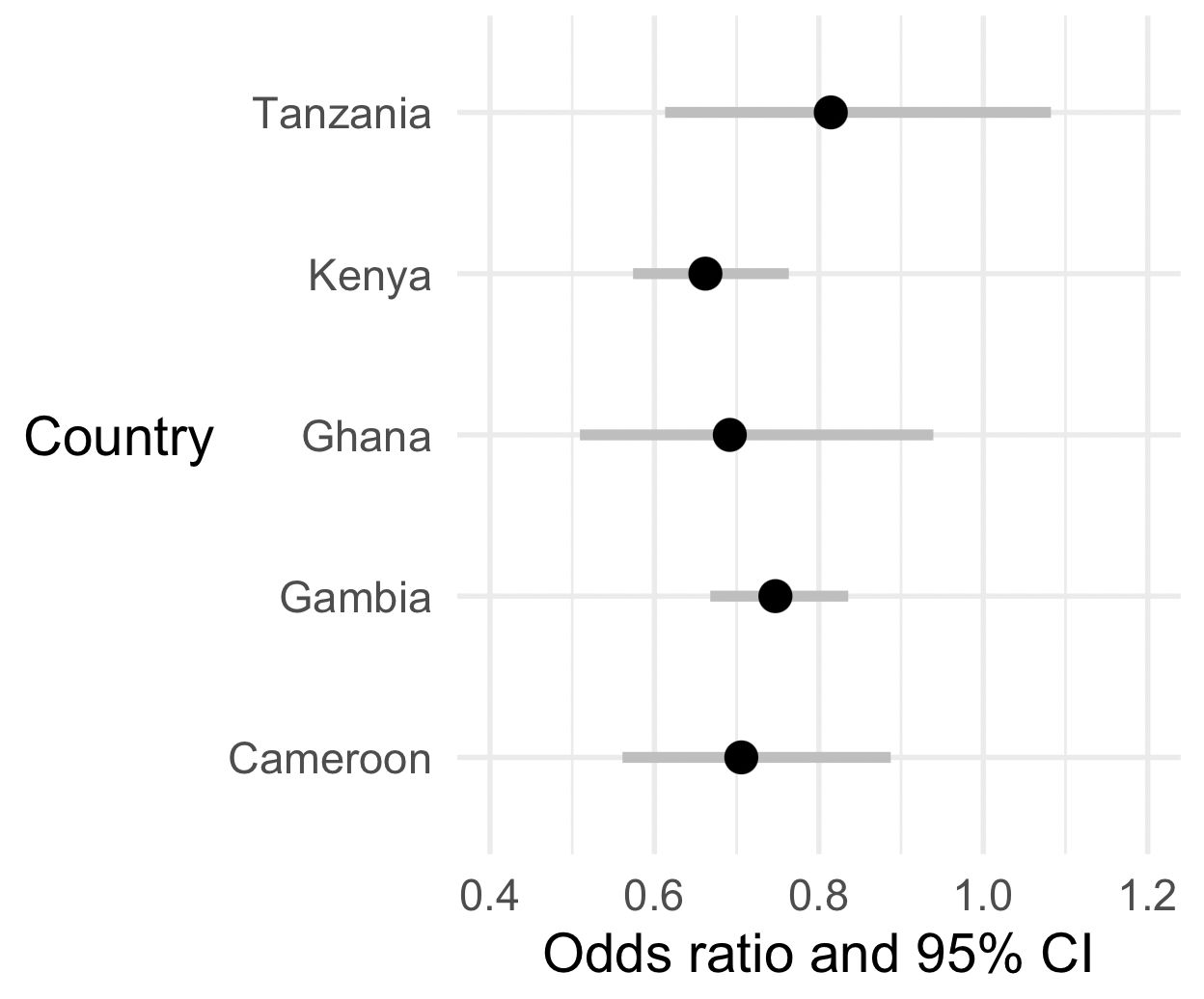
Nice? But not good enough, let's do two things to make it better!
First, let's add a vertical dashed line at (which corresponds to 'no association' on the odds ratio scale):
(
p
+ geom_vline( xintercept = 1, linewidth = 1.5, linetype = 2 )
)
The second thing is a bit more subtle. We would like the plot to be on the natural (odds ratio or relative risk) scale, as it is at the moment, because that's easiest to interpret.
But on the other hand the odds ratio scale is a bit weird for a plot! (It goes from to , and is multiplicative i.e. with corresponding to 'no association'.) So in that sense it seems better to have this plot on the log scale.
Luckily, we can have our cake and eat it by simply telling ggplot to plot on a log scale:
(
p
+ geom_vline( xintercept = 1, linewidth = 1.5, linetype = 2 )
+ scale_x_log10( limits = c( 0.4, 1.5 ))
)
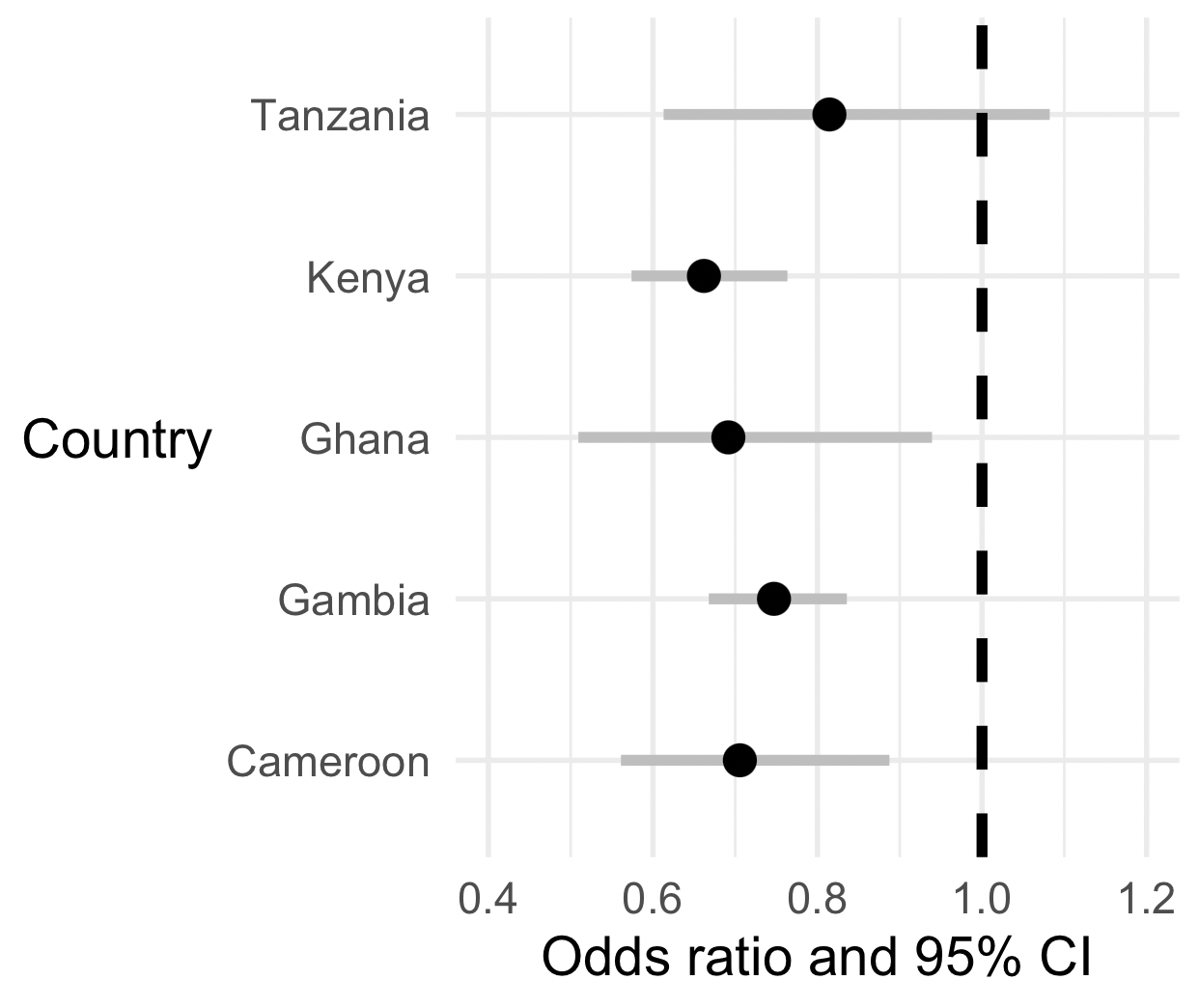
Nice!
What is your overall interpretation of these data?
Is O blood group likely associated with severe malaria across all these populations?
Congratulations - you've finished this part of the practical. Feel free to take a break! Or read more information below if you're interested.
Other topics
Understanding categorical covariates
If you add a categorical covariate, like ethnicity, into the regression:
fit = glm(
status ~ o_bld_group + ethnicity,
data = data %>% filter( country == 'Gambia' ),
family = "binomial"
)
summary( fit )$coeff
You will probably see that the output suddenly had lots of rows! Where did they come from?
What happens is that, under the hood, R transforms a variable like this:
ethnicity
EXNMY8743H ethnicity_1
HEQVB6322Z ethnicity_2
CGARJ3859K ethnicity_2
WQVWO2433A ethnicity_1
AJNTO3047P ethnicity_3
IMJJY8783Q ethnicity_2
...
...into a matrix like this:
ethnicity_1 ethnicity_2 ethnicity_3 ...
EXNMY8743H 1 0 0
HEQVB6322Z 0 1 0
CGARJ3859K 0 1 0
WQVWO2433A 1 0 0
AJNTO3047P 0 0 1
IMJJY8783Q 0 1 0
...
By default the first column gets removed (it corresponds to the baseline level) and then every other level gets its own parameter.
Logistic regression, case-control studies and relative risk
In the above you have hopefully identified the odds ratio as an estimate of the population relative risk. That's why we can interpret it as 'O blood group is associated with about a 30% reduction in risk of severe malaria'.
We will discuss in the lectures but here is a recap.
Suppose disease cases in the population come along at some frequency (for individuals with non-O blood group) and (for individuals with O blood group). The simplest way to measure association in the population is using the relative risk:
Key fact. The key fact discussed in lectures, is that in a case-control study if the controls are 'population controls' (i.e. sampled from members of the general population), the study odds ratio is an estimate of the population relative risk. The reason is that the above can be re-written using Bayes rule in terms of genotype frequencies in cases and population controls:
The four terms there are each estimated by can be estimated by taking a sample of cases and population controls, and measuring their genotypes.
How does this correspond to the logistic regression model? It turns out the logistic regression model we fit above is equivalent to this one in a 2x2 table:
If you 'do the maths' on this table (work out the odds ratio) you'll see that the odds ratio is juse .
Thus, even though the baseline parameter doesn't reflect the population risk of disease (because of the ), the odds ratio reflects the relative risk in the population.
In our case if the relative risk is about 0.7 - we could say O blood group is associated with about 30% reduction in risk of severe malaria.