Preprint on human-malaria genetic association across populations now out
Our preprint on the evolution of the sickle-associated variants is now out! Written in collaboration with Andre Python of Zhejiang University and with the support of my former postdoc Annie Forster, this is our attempt to understand the evolution of a set of interesting alleles in the Plasmodium falciparum genome. 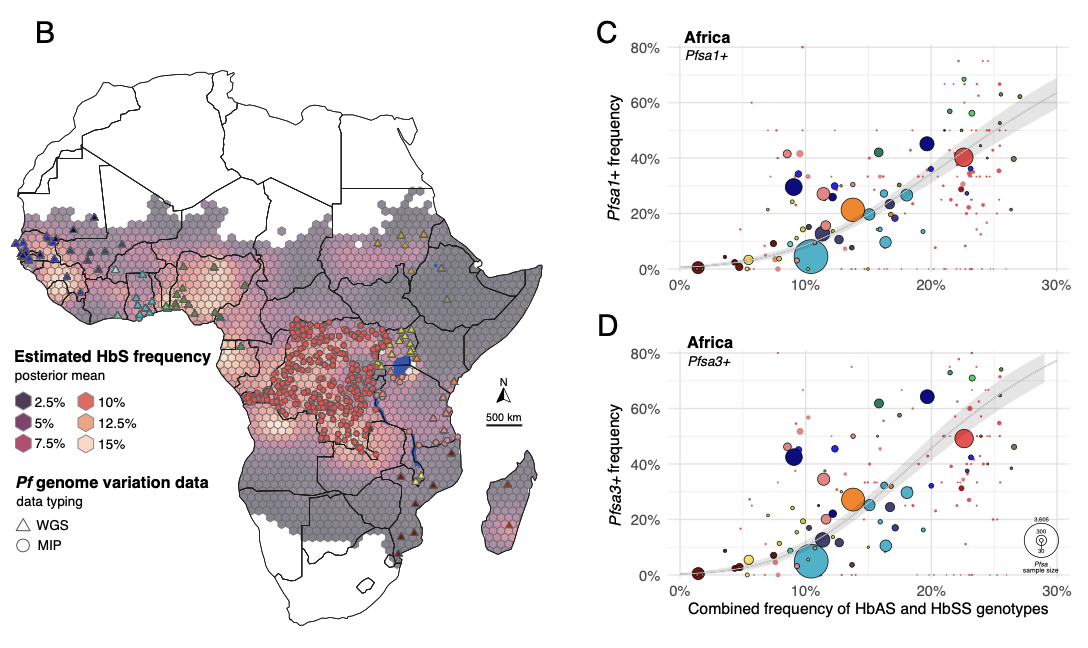
The alleles in question are the 'Pfsa+' alleles - genetic variants which seem to be able to overcome the protective effects of the sickle haemoglobin allele (HbS). But these variants also turn up in plenty of infections of people who don't carry HbS - they don't seem to carry much cost. So why are the variants still at intermediate frequencies in African populations, e.g. still polymorphic?
This paper provides a possible answer to that using three analyses. In the first analysis we show that the Pfsa+ allele frequencies are strongly correlated to host HbS frequencies across African populations - implying that the presence of HbS is selecting for the alleles, and suggesting there is in fact some negative cost to them in HbAA individuals. In the second analysis, we use a simulation model to provide a possible explanation - namely that these opposing fitness costs, when spread over a realistic geographical area, might lead to the alleles become balanced. If so they could stay at intermediate frequencies over long periods. 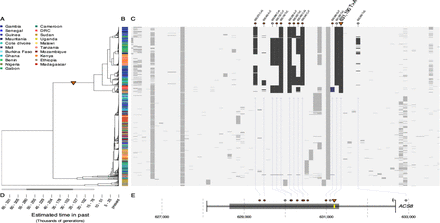
In the third analysis, we then examine the genealogies of the loci and find patterns that are - at least at two of the loci - also consistent with this idea. The Pfsa+ alleles seem to have a 'deep split' in the tree. The signal is somewhat subtle but is at least consistent with the idea they have arisen several thousand years ago, and then maintained since that time.
We completed this paper just as collaborators were