STITCH
Last update: January 4, 2017
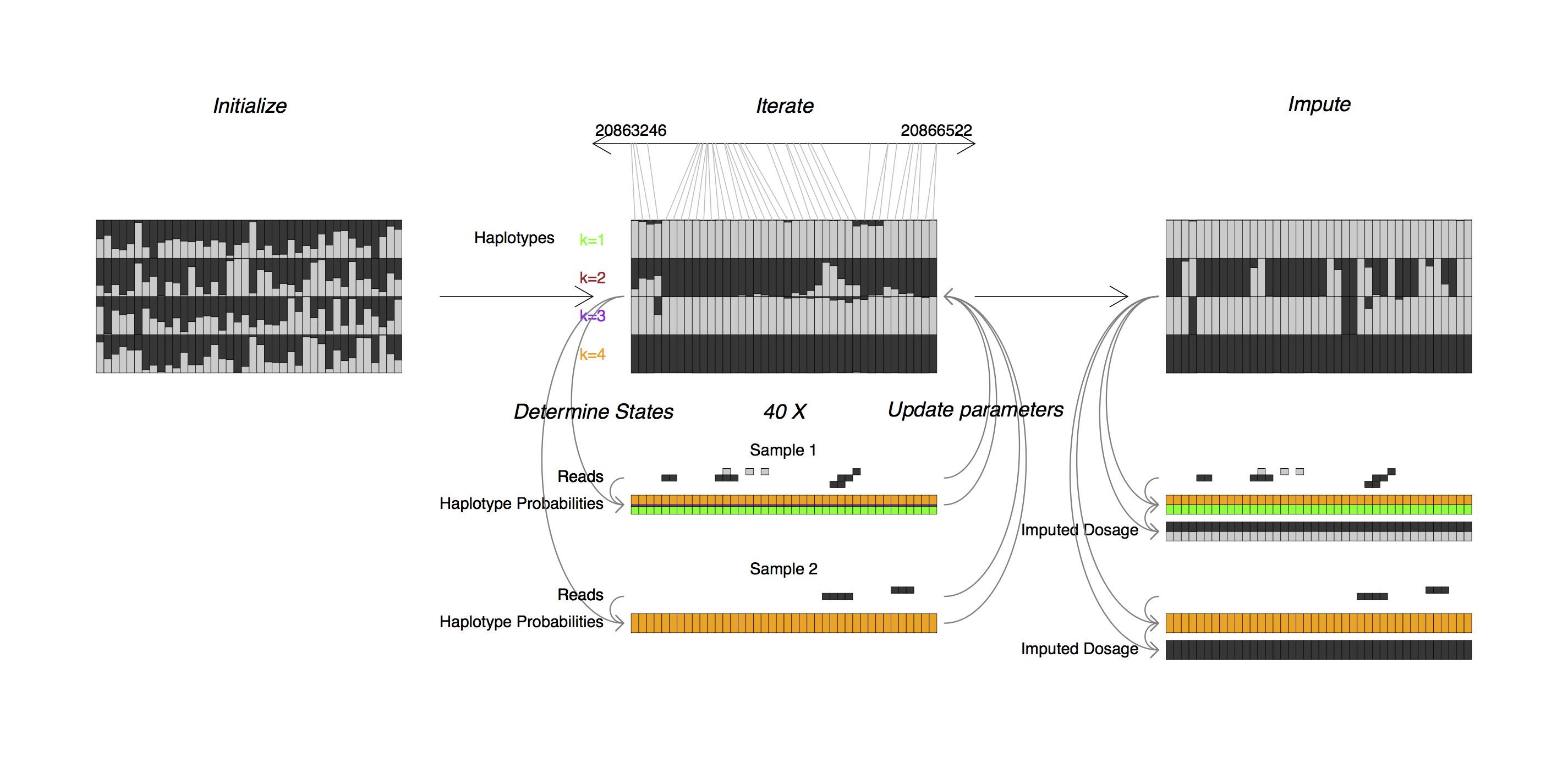
STITCH is an R program for reference panel free, read aware, low coverage sequencing genotype imputation. STITCH runs on a set of samples with sequencing reads in BAM format, as well as a list of positions to genotype, and outputs imputed genotypes in VCF format.
STITCH works by modelling each chromosome in the set of samples as a mosaic of K unknown founders or ancestral haplotypes. STITCH employs a hidden Markov model, whose parameters are sequentially updated using expectation maximization. Both steps are handled in a read aware fashion done without using external reference haplotype sets.
STITCH has been tested on low coverage mouse and human data. STTICH has been run on mouse genotype-by-sequencing (GBS) data, although it may encounter issues when the number of reads becomes close to 0. For guidance on parameter options (like K), please see the supplementary note in Davies et al. given below
License
STITCH is free for academic use. For commercial inquiries please contact Robert Davies (robertwilliamdavies at gmail dot com) and Simon Myers (myers at stats dot ox dot ac dot uk)
Changelog
- v1.2.4
- Enable C++11 compilation
- Fix bug where sample name from bam header was being grabbed from any line with @RG in it and not specifically lines starting with @RG
- v1.2.3
- Fix bug where the central SNP in a read was random from SNPs in read and not the central SNP by position among SNPs in the read as it ought to have been
- Fix bug where the final SNP from posfile wasn't being loaded from the sample BAMs and as a result not being imputed
- Fix bug where reads split into 3+ pieces were not being properly handled (e.g. long reads where sections map to multiple locations)
- Faster internal handling of cigar string
- v1.2.2
- Reduce RAM footprint when using reference panels
- Crash early if rsync is not in PATH
- Added option to override default VCF output name. See vcf_output_name
- Added unit tests under testthat framework
- v1.2.1
- Change internal system calls to reduce RAM usage
- Fix bug passing through variable to subfunction
- v1.2.0
- Can use reference panels in IMPUTE2 format. See example script and reference_* variables
- Can bundle together inputs to facilitate imputation of very large N. See inputBundleBlockSize
- Example human data provided to showcase STITCH functionality. See examples script
- v1.1.4
- Can work off CRAM files or BAM files. To use CRAM files, see cramlist and reference variables, or see examples script
- Changed GL as genotype likelihood to GP as genotype posterior probability in output VCF
- v1.1.3
- Changed R example script to work on provided example data
- Changed default downsampleToCov to 50 to reduce likelihood of overflow at high coverage SNPs
- Miscellaneous small fixes to BAM conversion script to better handle samples with very few reads
- Changed default region where reads from BAM are loaded (chrStart and chrEnd) to NA, to be inferred from posfile and the region to be imputed, rather than to grab reads from the whole chromosome
- Fixed ability to use high coverage validation samples (genfile) when using generateInputOnly and regenerateInput
- v1.1.2
- Fixed typo in header of outputted VCF
- v1.1.1
- Fixed bug where samples with no reads on a chromosome gave an old input format
- v1.1.0
- Changed default output to VCF (added option outputBlockSize to control how it is written)
- Removed two package dependencies
- Remove ability to write output to .gen format (remove outputGenFormat)
- Add change log to README and miscellaneous other changes
- v1.0.1
- Added ability to use soft clipped bases
- v1.0.0
Downloads
Complete working example
- Install R if not already installed. Install R dependencies parallel, Rcpp and RcppArmadillo from CRAN (using the "install.packages" option within R), and Rsamtools from BioConductor
- Download STITCH from above. Install by opening R and using install.packages, giving install.packages the path to the downloaded STITCH tar.gz file above
- Download example tar.gz from above and open using a command such as tar -xzvf
- Run STITCH. Open R, change your working directory using setwd() to the directory where the example tar.gz was unzipped, and then run STITCH(tempdir = tempdir(), chr = "chr19", bamlist = "bamlist.txt", posfile = "pos.txt", genfile = "gen.txt", outputdir = paste0(getwd(), "/"), K = 4, nGen = 100, nCores = 1). Once complete, a VCF should appear in the current working directory named stitch.chr19.vcf.gz
Contact
If you have any problems running STITCH please contact Robert Davies (robertwilliamdavies at gmail dot com)
Citation
If you use STITCH, please cite
Davies, R. W., Flint J, Myers S., Mott R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 48, 965-969 (2016)